In the past, for the Pdamicornis wound healing paper, I used STAR to align the 3’ RNAseq reads to the genome, not the transcriptome.
The notes I’m relying on for this round of analysis are from Dr. Natalia Andrade (https://github.com/China2302/transcriptomics-workflow) and Dr. Michael Studivan (https://github.com/mstudiva/tag-based_RNAseq/blob/master/). The reason for this is because I sequenced the Acer CCC samples with Natalia’s SCTLD Mote and Nova samples, while the temperature variability 2023 Acer and Pcli samples were sequenced following Michael’s protocols.
So far, because Michael’s stuff is all in python, I have had more success with Natalia’s pipeline. However, the QC and trimming steps are not super specific, and they both used cutadapt to trim Illumina adapters. Michael’s code had more specifics for trimming (i.e. specific base pair sequences) that are potentially more likely to be found in TagSeq generated reads. Natalia specifically trimmed the polyA tails because that is a problem in 3’ RNA seq.
But for alignment, there are many different possible tools to use, and there is also the question of whether to align to a genome or a transcriptome.
The UM CCS student mentors made a pros and cons list for aligning to a genome versus a transcriptome (https://github.com/ccsstudentmentors/tutorials/tree/master/RNA-Seq/Quantifying-RNA-Expression). I think it also depends on what is available for the species you’re analyzing.
Transcriptome – then use Bowtie.
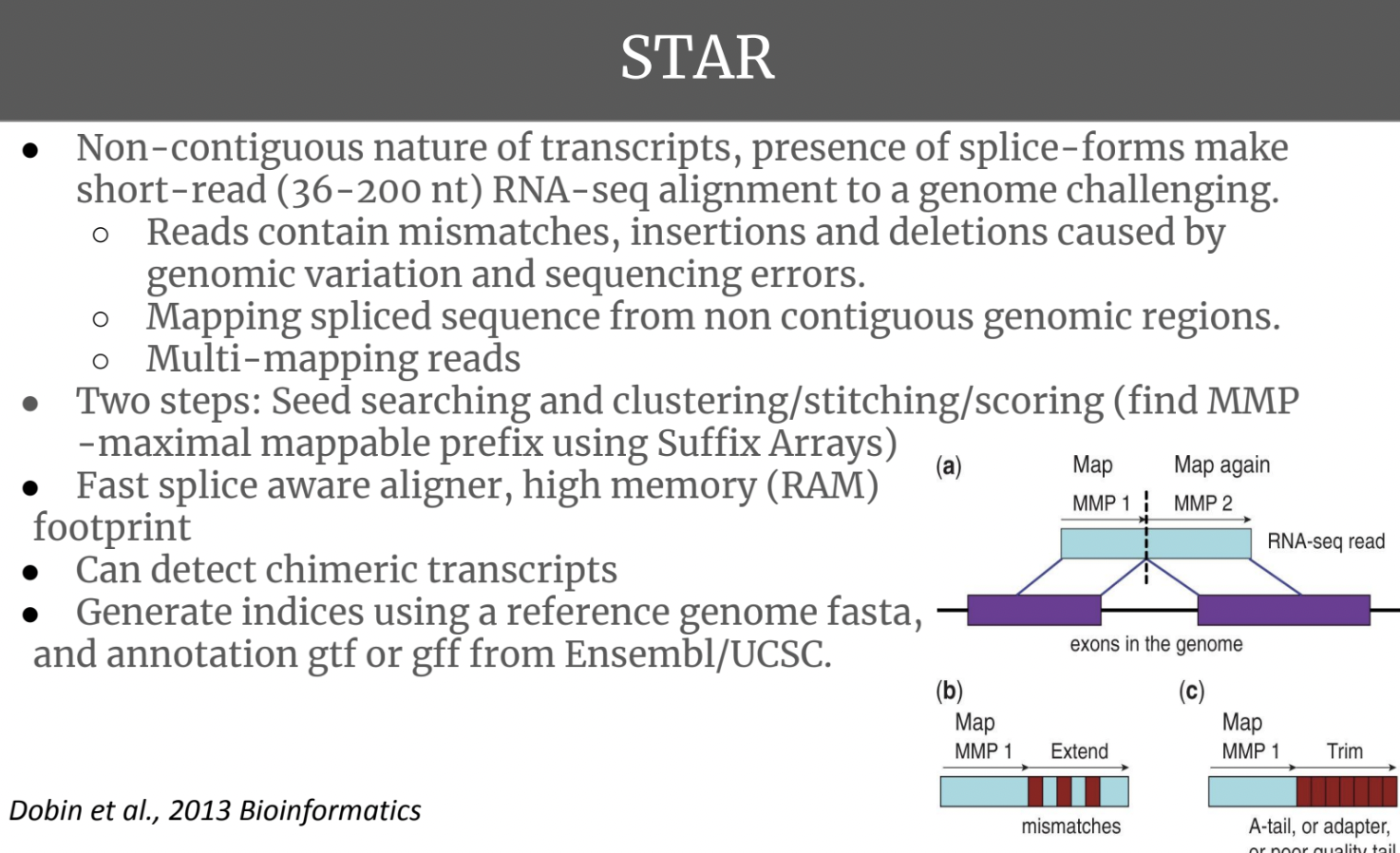
Genome – use STAR.
After talking to Dr. Kevin Wong, he said to just pick whichever gives the best alignment rate.
In terms of which tool to use, STAR seems to be the superior aligner:
- https://www.biostars.org/p/353946/